Diblock Copolymers (DBCs)
A diblock copolymer consists of an A-block of \( N_{A} \) segments and a B-block of \( N_{B} \) segments (figure 1), joined together at one end to give a linear chain with \( N = N_{A} + N_{B} \) total segments and composition fraction \( f_{A} = N_{A}/N \).

Figure 1 - A diblock copolymer with \( N_{A} \) segments in the A-block and \( N_{B} \) segments in the B-block.
The strength of interactions between A and B segments is controlled by the Flory-Huggins parameter, \( \chi \), which is inversely related to temperature (i.e., low \( \chi \) corresponds to high temperature). When the A-B interactions in a melt of DBCs become sufficiently unfavourable (i.e., \( \chi \) becomes large enough) they microphase-separate, forming periodically ordered structures with domains on the nanometer scale. The equilibrium structures include the classical lamellar (L), cylindrical (C) and spherical (S) phases, as well as the complex gyroid (G) and Fddd phases (see figure 2).

Lamellar (L)

Cylinder(C)

Spherical (S)

Gyroid (G)
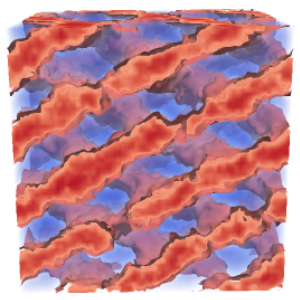
Fddd (Fddd)
Self-consistent Field Theory (SCFT)
Imagine looking at a single DBC in a melt. It has complicated interactions with all the other polymers in the system and capturing them all is a difficult mathematical and computational challenge. SCFT addresses this problem by replacing the interactions that a polymer has with other chains by mean fields.
Monodisperse Phase Diagram
SCFT has been enormously successful in the study of DBCs, producing the famous equilibrium phase diagram for a monodisperse melt shown in figure 3. It displays the various ordered phases, which are separated from the disordered phase by the order-disorder transition (ODT). It is also worth noting that the phase diagram is symmetric about \( f_{A} = 1/2 \).

Figure 3 - The self-consistent field theory phase diagram for diblock copolymer melts.

Figure 4 - A schematic representation of the effect of fluctuations on the SCFT phase diagram in figure 3.
Limitations of SCFT
Unfortunately, the mean-field nature of SCFT means that it doesn't include the fluctuation effects that are inherent in real statistical systems. As a result, we are interested in fluctuation corrections to figure 3, which are controlled by the invariant polymerisation index, \( \bar{N} = a^{6}\rho_{0}^{2}N \), where \( \rho_{0} \) is the segment density and \( a \) is the statistical segment length.
SCFT corresponds to the case where \( \bar{N} = \infty \), with fluctuations increasing as \( \bar{N} \) is reduced. The main effects of fluctuations on the monodisperse phase diagram in figure 3 are that the ODT gets shifted upward, and we get direct transitions between the disordered and ordered phases. For example, there will be a direct transition beween gyroid and disorder as indicated schematically in figure 4.
Monodisperse Phase Diagram from Experiments
In 1994, Bates et. al. performed experiments to produce monodisperse phase diagrams for three different DBC chemistries: PE-PEP (\( \bar{N}=27 \times 10^{3} \)), PEP-PEE (\( \bar{N}=3.4 \times 10^{3} \)) and PI-PS (\( \bar{N}=1.1 \times 10^{3} \)). Each of the phase diagrams exhibited the spherical, cylindrical and lamellar phases, but no gyroid or Fddd. There was, however, a region of another phase known as perforated lamellar in the region where SCFT predicts gyroid. Indeed, later experiments by Hajduk et. al. and Vigild et. al. showed that perforated lamellar is actually a long-lived metastable state that eventually converts to gyroid. Even more recent experiments by Wang et. al. and Takenaka et. al. have also confirmed the presence of Fddd on both sides of the phase diagram.
Whilst the experiments and SCFT phase diagrams exhibit a lot of good qualitative agreement, the significant differences are due to the effects of fluctuations near the ODT in the experiments: the shifting up of the ODT and direct transitions between disorder and the ordered phases.